

What are ciliopathies?
Ciliopathies are complex disorders caused by genetic mutations which result in defective or dysfunctional cilia in many organs of the human body.
Alström Syndrome is a very rare recessively inherited condition which affects the metabolism of many major organs, particularly the heart, lungs, kidneys and liver.
Because the condition unfolds gradually from birth and the different manifestations vary from individual to individual, correct diagnosis is often delayed leading to suboptimal treatment and a failure to anticipate future developments. There are presently just over 1000 cases known worldwide and approximately 100 in the UK although the history of poor diagnosis referred to above almost certainly hides many more.
Alström Syndrome is characterised principally by a number of key conditions:
- Retinal degeneration (Rod Cone Dystrophy, Nystagmus and Photophobia)
- Sensorineural hearing loss (ranging from a mild, moderate to severe loss
- Obesity
- Insulin resistance
Additional features can include:
- Cardiomyopathy (poor cardiac function where the heart muscle is weakened and enlarged
- Type 2 diabetes
- Renal and hepatic dysfunction (affecting the kidneys and liver)
- Hypertriglyceridaemia and tryglycerides (elevation of fatty substances found in the bloodstream)
How is Alström Syndrome diagnosed?
Dilated cardiomyopathy is often detected in young babies and often leads to premature death. Kidney failure may also occur. During childhood, patients suffer deteriorating eyesight which may include nystagmus and photophobia which often leads to blindness. Hearing loss and Type 2 diabetes is also common.
Find out more about Alström Syndrome
Useful links
Bardet-Biedl Syndrome (BBS) is a rare, recessively inherited complex disorder that involves many body systems. Mutations in more than 20 different genes encoding proteins involved in cilia formation, maintenance and signalling can cause BBS. Genetic screening is a major part of the diagnostic pathway. There is at present no cure for Bardet-Biedl syndrome. Patients have access to NHS specialised multi-disciplinary clinics held in four centres in London and Birmingham. Bardet-Biedl Syndrome UK are third-sector partners in this service.
The incidence is estimated at 1 in 100,000 births.
Diagnosis
Beales et al (1999 and 2001) suggest that the presence of four primary features or three primary features plus two secondary features is necessary for a clinical diagnosis of Bardet-Biedl syndrome.
Primary:
- Rod-Cone Dystrophy
- Obesity which usually begins in childhood and increases in severity with age
- Extra fingers and/or toes (polydactyly) and/or partially fused digits (syndactyly)
- Kidney abnormalities, with a minority affected by renal failure
- Developmental delay, speech delay and learning difficulties
- Hypogonadism in males
Secondary:
- Speech delay/disorder
- Developmental delay
- Brachydactyly
- Polyuria/polydipsia
- Ataxia
- Poor co-ordination
- Diabetes mellitus
- Left ventricular hypertrophy
- Hepatic fibrosis
- Hypertonia
- Hearing loss
Impact of BBS
- Complex syndrome, delayed diagnosis is common
- No treatment for rod-cone dystrophy, correct early diagnosis vital for future development
- Obesity is difficult to treat, lifelong commitment to healthy diet and exercise necessary
- Learning difficulties and speech problems need early intervention for successful outcome
- Low mood, anxiety, anger and poor emotional control commonly affect adults and young people with BBS
Find out more about Bardet-Biedl Syndrome
Useful links
Jeune Syndrome, also called short-rib thoracic dysplasia, is a very rare (estimated incidence of 1 per 200,000) group of recessively inherited ciliary .
Primary features
- Narrow/ small thorax due to short ribs - this causes underdevelopment of the lungs in utero.
- Respiratory distress at birth and in infancy which improves later in life due to postnatal rib growth.
- Shortening of the arms, legs, fingers and toes (brachydactyly) may be present.
- Cone shaped epiphyses (ends of the bones) often become visible in x-ray images of the hands after the first year of life.
- Sometimes, extra fingers and toes (polydactyly) are present.
- Rarely, retinal disease such as retinitis pigmentosa or rod-cone dystrophy. These conditions cause impaired vision and may progress to blindness.
- Renal disease due to polycystic or nephronophthisis-like kidney symptoms which may progress into renal insufficiency.
- Mild liver dysfunction is often reported which only rarely seems to progress into more severe liver disease.
Impact of JATD
- Lethality occurs in 20-60% of all cases, mainly during the first 1-2 years of life. This is most often due to respiratory problems resulting from a narrow ribcage. Some patients seem to "grow out" of the rib phenotype later in life. Nevertheless, the narrow ribcage can cause mechanical lung compression during pregnancy in female patients. Patients also might more frequently develop scoliosis and hip dysplasia.
- Mutations in different genes are linked to variability of clinical features, for example IFT140 mutations are often associated with early renal failure
- There is no curative therapy available for JATD to date, therapeutic options are limited to supportive measures such as mechanical ventilation or thoracic expansion surgery in severe cases and appropriate treatment of respiratory infections. Renal disease should be diagnosed early and patients should receive appropriate supportive treatment such as dialysis and renal transplantation. Retinal disease should be excluded and/or monitored via regular ERG (electroretinogram) examinations.
- Extra fingers and toes can be removed surgically if they cause functional problems.
Find out more
Useful links
Joubert Syndrome is a rare developmental disorder affecting mainly the brain but this might be accompanied by renal and/or retinal symptoms. Mutations in several genes associated with cilia can cause Joubert Syndrome which are inherited often in a autosomal-recessive manner but x-chromosomal-recessive inheritance also occurs.
Primary features
- Abnormalities of the brain, mainly the cerebellum-part lead to low muscle tone at birth, developmental delay and there may also be breathing abnormalities and movement disorders.
- Brain scans such as magnetic resonance tomography (MRI) show a characteristic feature of the disease, named the “molar tooth sign”.
- Renal disease, usually in the form of Nephronophthisis, may also occur and can lead to renal insufficiency.
- Babies can be observed to have poor sight at birth which can improve when the body learns to co-ordinate movement and muscle tone develops. Mystagmus is common.
- Extra fingers and toes (Polydactyly) are sometimes observed.
Impact
- There is a broad phenotypic spectrum of the brain phenotype among patients ranging from developmental delay and slight movement problems to severe brain defects including open brain that might be incompatible with life.
- Breathing problems are due to cerebral mis-regulation rather than to primary problems in the respiratory tract. Newborn babies sometimes require a tracheostomy to help with breathing difficulties.
- No cure is currently available for Joubert Syndrome, only supportive therapy.
Find out more
Useful links
Nephronophthisis (NPHP) is the most common genetic cause of chronic kidney disease within the first three decades of life.
Presentation may occur during infancy but more typically in late childhood with progressive renal failure manifesting during early puberty.
Primary features of Nephronophthisis
Ultrasonographic features demonstrate normal size kidneys with loss of cortico-medullary differentiation and increased echogenicity.
Histologically, NPHP kidneys are characterized by the presence of cortico-medullary cysts, tubular basement membrane disruption and tubulointerstitial nephropathy.
Inherited in an autosomal recessive mode, NPHP is genetically heterogeneous with at least 17 genes currently implicated which account for only about 30% of cases. NPHP is a ciliopathy disorder because similarly to the proteins involved in polycystic kidney disease, the nephrocystins (NPHP associated proteins) have all been localized to primary cilia, basal bodies and centrosomes. Many different ciliopathy patients show symptoms of NPHP.
Impact and symptoms of NPHP can include
- Polyuria (passing large volumes of urine frequently)
- Polydipsia (excessive drinking)
- Secondary enuresis (wetting)
- Anaemia
- NPHP may also have extrarenal manifestations, such as liver fibrosis, situs inversus, or cardiac malformations.
Find out more about Nephronopthisis
Polycystic Kidney Disease (PKD) refers to a range of genetic diseases which are a common cause of kidney failure in children and adults.
There are two main forms of PKD:
- Autosomal Dominant Polycystic Kidney Disease (ADPKD)
- Autosomal Recessive Polycystic Kidney Disease (ARPKD)
What is ADPKD?
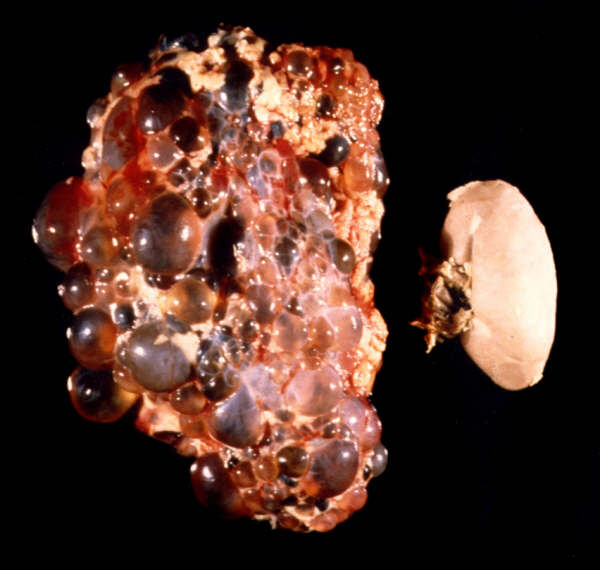
- The world’s most common inherited life-threatening condition
- The most common inherited kidney disease
- Characterised by fluid-filled cysts which develop, enlarge and multiply in both kidneys causing progressive renal failure
- Multi-system condition, affecting other organs, such as the liver, pancreas, spleen, brain, intestines
- Prevalence: between 1 in 500 and 1 in 1000 – approx 12.5 million worldwide, 70,000+ in UK
- Men and women equally affected with no apparent racial bias
- Two genes: PKD1 and PKD2 with differing outcomes
- Typical age of onset: 30s to 50s
- Results in end stage renal failure in 50% of patients
- Accounts for 8-10% of patients on RRT (renal replacement therapy)
- Currently incurable
What is ARPKD?
- Rare form of PKD
- Incidence: 1 in 20,000 live births
- In utero: sometimes fatal; babies have enlarged kidneys and little amniotic fluid; there is failure of the lungs to fully develop; and there may be deformities of spine and limbs
- In newborn: 30%-50% die at birth or shortly thereafter; ~40% of survivors have respiratory failure needing ventilation
- 12% of ARPKD children develop chronic lung disease
- Some may require a nephrectomy
- Children usually have hypertension and problems with salt and water balance
- Kidney function is usually poor; infections are common
- Kidney failure often happens by age 30
- Liver abnormalities are common, primarily Congenital Hepatic Fibrosis
Find out more about PKD
Useful links
Visit PKD International for a list of PKD patient groups worldwide
Primary Ciliary Dyskinesia (PCD) is the only known disorder of the motile cilia. However, dysfunction of motile cilia is implicated in several primary cilia ciliopathies - but these links are not yet well established. PCD is an autosomal recessive disorder which presents with upper and lower respiratory tract infection, and affects the lungs, sinuses and ears. Other features of PCD reflect dysfunction of cilia motility outside the airways, including subfertility, hydrocephalus and body laterality (left-right axis) defects. Some rare cases have retinal and neurological problems.
The incidence is 1 in 7500 . However, the incidence can be as high as 1 in 2,500 in the Asian population and other areas where consanguineous marriages are prevalent.
Recent studies have begun to locate PCD genes scattered throughout the genome, making it a heterogeneous condition. More than 50 different genes have been found to cause PCD, either with recessive inheritance and more rare X-linked-recessive inheritance.
PCD affects both sexes and all populations. It is managed by physiotherapy and targetted antibiotics.
Symptoms of PCD
- Chronic respiratory infection
- Bronchiectasis
- Progressive loss of lung function
- Nasal problems
- Sinus disease
- Hearing loss
- Male infertility
- Hydrocephalus
- 50% laterality defects
- Heart defects 6-17 %
- Ectopic pregnancy
Impact of PCD
Affected individuals will experience lifelong chronic lung disorders and will typically need:
- Twice daily physiotherapy
- Targeted antibiotics either orally or intravenously
If PCD is not diagnosed, there is a risk of permanent lung damage.
Hearing and fertility problems are common.
How is PCD diagnosed?
PCD arises from cilia dysmotility associated with cilia structural defects or lack of cilia, that can usually be detected using specialised microscopy. Screening tests for PCD include nasal nitric oxide and tests of ciliary motility by high speed video imaging of nasal cells. Specific diagnosis requires examination of cilia by light and electron microscopy, with epithelial culture in doubtful cases. Genetic testing is increasingly used to assist PCD diagnosis especially since up to a fifth of cases may not show any obvious structural cilia defects.
Find out more about Primary Ciliary Dyskinesia
Useful links
Retinitis pigmentosa (RP) is the name given to a large group of inherited diseases of the retina that all lead to a gradual progressive loss of vision (Inherited Retinal Dystrophies). The term encompasses many named conditions including retinal ciliopathies such as classic retinitis pigmentosa, cone-dystrophy, cone-rod dystrophy and Leber congenital amaurosis. Difficulties with night vision and peripheral vision are usually the first things that are noticed. Later, detailed and colour vision are affected. The age at which symptoms start is variable and the rate of deterioration may vary but typically night blindness and loss of peripheral vision is followed by tunnel vision. Ultimately this window on the world may shrink and close up altogether.
RP is caused by flaws in at least 300 genes, some of which have not yet been fully characterised or even identified. Research is progressing, there is now one gene-specific therapy, called Luxturna, available on the NHS; this is for RP caused by faults in the RPE65 gene. However, this is not a ciliopathy gene, but instead is involved in the visual cycle.
Primary features
- Variable patterns and rates of retinal degeneration (sight loss).
- 1/3,500 approximate incidence.
- Syndromic forms of RP involve other disabilities or difficulties.
- Affects all ages, races and both sexes.
- A major cause of sight loss in people of working age and children.
- Currently only one treatment is available for a very specific gene fault (although this is not a ciliopathy gene however clinical trials are underway for gene therapy, use of stem cells, and other prospective treatments
Impact of RP
- Progressive sight loss of variable degree, pattern and rate.
- Practical assistance and lifestyle changes are necessary to cope with reduced vision or blindness.
- Significant emotional impacts in RP families, especially where there are multiple generations of people affected.
Find out more
http://www.ncbi.nlm.nih.gov/pmc/?term=retinitis+pigmentosa
Useful links
Usher syndrome is a rare autosomal recessive genetic condition that affects hearing, vision and balance.
Types of Usher syndrome
There are three clinical types of Usher syndrome – Type 1, 2 and 3. Type 1 and 2 are the most common. Symptoms Types 1 or 2 are present at birth however individuals affected by type 3 develop symptoms in childhood.
Type one
- Hearing: People with Usher type one are profoundly deaf from birth and have balance problems. Most use sign language as their primary means of communication. The hearing loss often remains stable throughout a person's life and is generally not helped by hearing aids. However, children may benefit from cochlear implants, thereby allowing them to develop speech.
- Vision: Most children with Usher type one usually begin to develop retinitis pigmentosa-related vision problems between the ages of 8-12 years old, with vision problems first noticed at night, followed by increasing difficulty with side (peripheral) vision. Visual problems are progressive.
- Balance: The balance problems mean that children may be late in sitting up and walking.
Type two
- Hearing: Children with Usher two are born with moderate to severe hearing impairment and normal balance. The severity of hearing impairment varies, but many, if not most, children can benefit from hearing aids. Children are likely to use speech to communicate.
- Vision: The visual problems related to retinitis pigmentosa (RP) tend to progress more slowly than in Usher one and also tend to begin later, usually in late teenage years, or may not even begin until the person is in their 30s or 40s. The RP is variable from person to person, so it is impossible to predict how much sight someone might have at any given stage in life.
The person with Usher two is faced with continually adapting to two changing senses and the impact a change in one sense has on their ability to use the other.
Type three
- Hearing: Children born with Usher three have normal hearing. Hearing worsens over time and may progress to profound hearing loss. However, the rate at which hearing is lost can vary between individuals, even within the same family. Children may develop noticeable hearing problems by their teens and usually are using hearing aids by mid to late adulthood.
- Vision: Vision problems usually begin during teenage years, starting with night blindness. The sight problems are more variable and this can mean the central vision is affected earlier. As with Usher two, the onset and progression of RP is variable from person to person.
- Balance: Some people with Usher Type 3 will have near normal balance but some may develop problems later on.
Identified genes
So far at least ten have been identified that cause the disorder. They are:
- Type one Usher syndrome: MY07A, USH1C, CDH23, PCHD15, SANS
- Type two Usher syndrome: USH2A, ADGRV1 (previously called VLGR1), WHRN
- Type three Usher syndrome: USH3A, HARDS
Treatment
There is currently no curative treatment for Usher syndrome. Supportive treatment is directed towards symptoms that have developed. Research towards possible genetic therapies aiming to slow the progression of visual loss associated with RP is ongoing. Some research has suggested that Vitamin A supplementation may be beneficial.